PFMEA for Manufacturing: When and How to Use It

If your last process audit ended with the auditor pulling up a five-year-old PFMEA spreadsheet, asking why a high-severity failure mode still had a Risk Priority Number of 84 and no documented mitigation, you already know the problem. Process FMEA is one of the most widely used risk tools in regulated manufacturing and one of the most consistently misapplied. Many teams inherit a PFMEA template, calculate RPNs out of habit, and update the document only when a deviation forces the issue. That version is a compliance artifact, not a working risk control.
This guide is for the engineer who needs PFMEA to do real work: drive validation scope, justify in-process controls, and survive audit scrutiny under the current AIAG-VDA handbook. It covers what PFMEA actually is, where it sits relative to DFMEA and other manufacturing FMEAs, when regulators expect it, how the AIAG-VDA Action Priority method replaced the old RPN approach, a worked PFMEA example end to end, and the mistakes that flag during inspections.
What PFMEA actually is
A Process Failure Mode and Effects Analysis is a structured method for identifying how a manufacturing process can fail to deliver its intended output, estimating how serious each failure would be, how likely it is to occur, and how likely it is to escape downstream detection, and then ranking those failure modes so the team can apply prevention and detection controls to the ones that matter most. PFMEA is a process-level analysis. Design FMEA looks at the product. Process FMEA looks at how the process produces the product.
A useful working definition: the PFMEA is the document a new process engineer can read to understand what your team has decided is risky about this process, what controls are in place to keep those risks acceptable, and what evidence shows the controls are working. If a PFMEA does not answer those three questions clearly, it is not doing its job.
PFMEA outputs are also the natural input to several other documents. It informs the Validation Master Plan by surfacing which process steps need the most validation coverage. It feeds the control plan by identifying the process and product characteristics that need in-process monitoring. It feeds the process validation strategy by clarifying which critical process parameters and critical quality attributes the validation program has to demonstrate control over. And it informs change control, because a documented PFMEA gives a change review team a defensible way to assess whether a proposed change shifts the residual risk profile of the process.
PFMEA is risk-based, meaning the depth of analysis on any given step is proportional to the risk that step presents. It is living, meaning it changes when the process changes. And it is integrated with the QMS, meaning its findings drive actions that get tracked in the systems the rest of the organization uses. A PFMEA that fails any of those three properties is what auditors find sitting in a folder no one has opened since the last submission.
PFMEA, DFMEA, and the other manufacturing FMEAs
The FMEA family is wider than most engineers realize, and confusing them in front of an auditor is a small but real credibility hit.
Design FMEA evaluates how a product design can fail to meet its requirements. The failure modes are functional. The team that owns it is engineering. The output drives design verification testing and design controls under 21 CFR Part 820 Subpart C and ISO 13485 clause 7.3.
Process FMEA evaluates how the manufacturing process can fail to make the product as designed. The failure modes are process modes such as wrong temperature, missed weld, contaminated raw material, missed inspection step. The team that owns it is manufacturing engineering or process engineering. The output drives the control plan, the validation strategy, and the in-process inspection plan.
System FMEA evaluates how an integrated system can fail at the system level, typically before any module-level design has stabilized. It is most common in automotive and aerospace and less common in pharma, biotech, and medical device manufacturing.
Machinery FMEA, sometimes called MFMEA, focuses on the equipment itself rather than the process the equipment runs. Failure modes are equipment failures: motor stall, sensor drift, hydraulic leak. MFMEA output usually drives preventive maintenance scope and reliability engineering programs.
Software FMEA evaluates how a software function can fail and what the downstream effect on the process or product is. It is increasingly relevant as more manufacturing equipment ships with embedded software and as more processes rely on connected MES and SCADA layers. Categorizing that software under the GAMP 5 framework decides how much validation each component carries.
For many regulated manufacturing programs, DFMEA and PFMEA are common inputs to design transfer and validation when product and process risk justify them. MFMEA is usually optional but valuable for equipment-heavy processes. SFMEA at the system level is rare outside automotive. Software FMEA is increasingly used for processes that include connected equipment with embedded firmware.
When PFMEA is required versus when it is good practice
PFMEA does not appear by name in 21 CFR Part 820 or in the updated FDA Quality Management System Regulation. What the regulations do require is risk management, and PFMEA is the most widely accepted method for satisfying that requirement at the process level.
For medical device manufacturers in the United States, the QMSR took effect February 2, 2026 and incorporates ISO 13485:2016 by reference. ISO 13485:2016 clause 7.5.6 requires that processes whose output cannot be fully verified by subsequent monitoring or measurement be validated, and clause 7.1 requires risk management throughout product realization. ISO 14971:2019 is the core medical device risk management standard. PFMEA can complement the manufacturing and process-control portion of that broader risk framework where process failure could affect device safety, quality, or compliance. The pre-QMSR QS Regulation expressed similar expectations in 21 CFR 820.30(g) for design validation and 21 CFR 820.70(a) for production controls. Teams whose documentation predates the transition will still reference those clauses.
For pharmaceutical manufacturers, ICH Q9(R1) is the umbrella guidance on quality risk management. ICH Q9(R1) does not require PFMEA specifically, but it lists FMEA and FMECA among the recognized risk assessment tools and frames them as appropriate for evaluating process risks. EU GMP Annex 15 §3 expects qualification activities to be risk based, and Annex 1 §3 for sterile products is similarly explicit. For APIs, ICH Q7 should be applied alongside ICH Q9 and ICH Q10. Risk management can inform validation and change decisions, but PFMEA is a method choice rather than a named ICH Q7 requirement.
For automotive, AIAG-VDA FMEA is commonly required by OEMs and Tier suppliers. Aerospace programs more often follow customer-specific or sector-specific risk methods. The 2019 AIAG-VDA FMEA Handbook is the current reference and is the method many teams outside automotive borrow from, because the clearer Action Priority framework and the cross-functional team structure travel well into pharma, biotech, and medical device manufacturing even when those teams are not formally required to use it.
So the practical answer to the "is PFMEA required" question is: no regulation requires PFMEA by name in pharma or medical device manufacturing, but the underlying risk management expectation is real and PFMEA is the tool most teams use to satisfy it. Programs that skip PFMEA in favor of an unstructured risk discussion often end up reconstructing equivalent risk logic during audit preparation or audit response.
Why AIAG-VDA replaced RPN with Action Priority
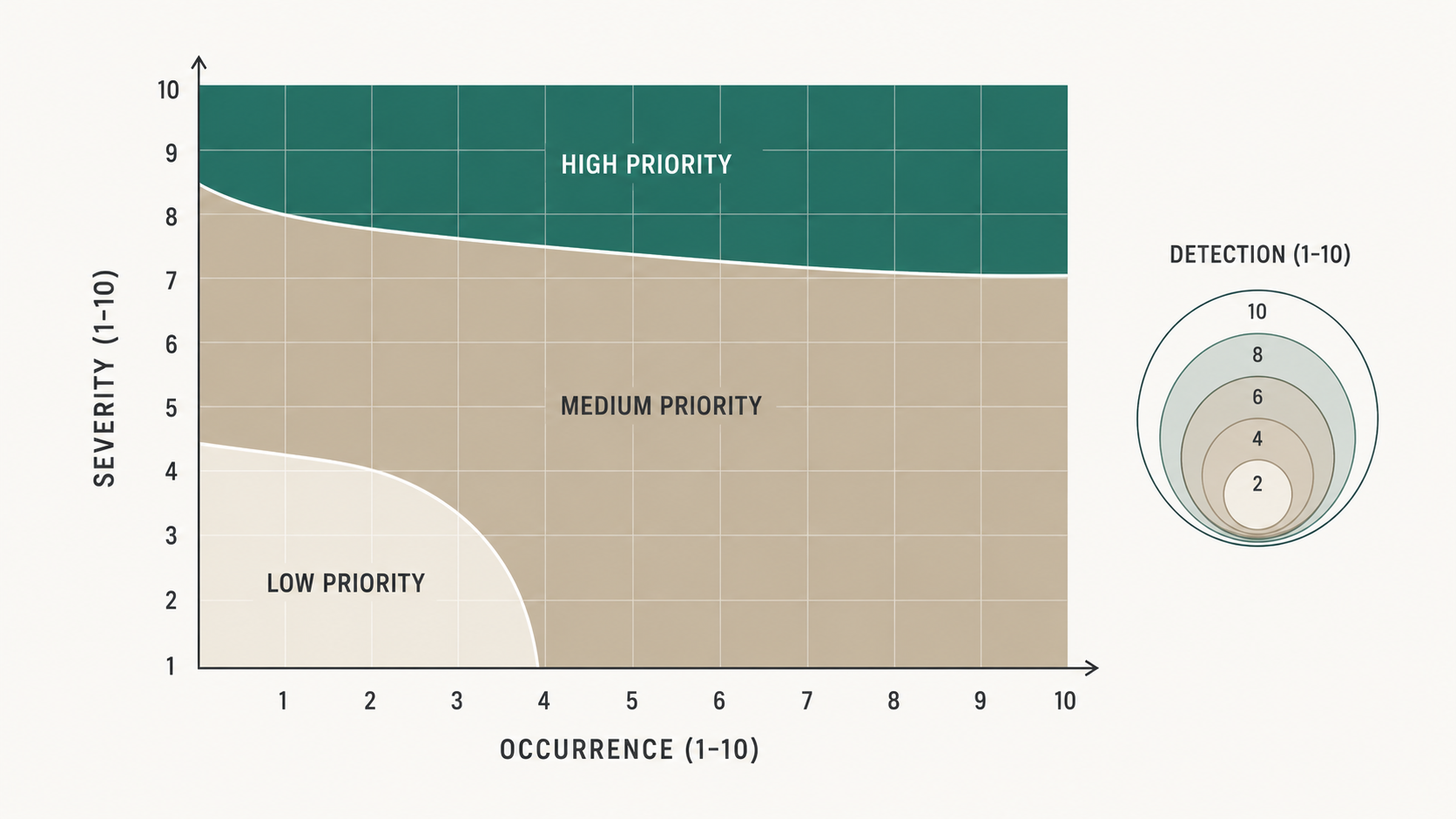
The 2019 AIAG-VDA FMEA Handbook made one substantive change that quietly transformed how PFMEAs get used in audit context. The old AIAG fourth edition ranked failure modes by Risk Priority Number, calculated as Severity times Occurrence times Detection. The handbook replaced RPN with Action Priority, a three-bucket classification of High, Medium, or Low that is determined by reading S, O, and D together against a published table rather than multiplied into a single number.
The reason for the change is that RPN treats all three dimensions as commutative. An RPN of 100 produced by S=10, O=2, D=5 looks the same as an RPN of 100 produced by S=2, O=10, D=5. The first scenario describes a catastrophic failure with a robust detection control. The second describes an annoyance that happens constantly. The actions a process engineer should take are completely different, but RPN gives them the same number and the same priority. Many teams responded by setting an RPN threshold (a common one was 100), working everything above the threshold, and ignoring everything below it. That created a perverse incentive: lower one of the three numbers, regardless of which one, and the failure mode falls below the threshold.
Action Priority encodes the asymmetry directly. A failure mode with very high severity is High priority almost regardless of occurrence and detection, because catastrophic failures need design changes even when they are rare and detectable. A failure mode with moderate severity and high occurrence is also High priority, because frequent failures consume manufacturing capacity even when their per-event consequence is bounded. A failure mode with low severity, low occurrence, and high detection is Low priority and the team can document and move on.
The handbook also tightened the Severity, Occurrence, and Detection rubrics so the rating descriptions reference observable manufacturing realities rather than abstract probabilities. Severity descriptions reference the impact on the end user, on regulatory compliance, on the customer, and on the plant. Occurrence descriptions reference how many parts per million the failure has been observed in similar processes or in historical data for this process. Detection descriptions reference how the control catches the failure, with prevention controls ranked separately from detection controls.
The practical effect is that an AIAG-VDA PFMEA is harder to game and easier to read. An auditor scanning a PFMEA can see at a glance which process steps the team has decided need design-level attention, which need stronger controls, and which the team has accepted residual risk on. The shift is from a number that summarizes risk to a categorical priority that triggers a specific action class.
Building a PFMEA end to end
A defensible PFMEA has a consistent build sequence regardless of process complexity. The categories below appear in nearly every audit-ready PFMEA we have read.
Step 1: Define scope and process boundaries
State the process under analysis, the start and end boundaries, the product family or families it produces, the production location, and the cross-functional team that participated in the analysis. A PFMEA whose scope is unclear is a PFMEA whose findings are unclear. Include a one-line statement of what the PFMEA does not cover and where coverage continues, for example "incoming material qualification is covered in the supplier quality risk assessment; this PFMEA begins at material release to production."
Step 2: Decompose the process into steps
Break the process into the granular steps a manufacturing engineer would recognize from the work instructions. A reasonable rule of thumb is one PFMEA row per process step, with sub-rows for steps that have multiple failure modes. Aim for steps that a single operator or a single automated cycle would perform. Steps that are too coarse hide failure modes; steps that are too granular create a document so long it never gets read.
Step 3: Identify failure modes for each step
For each step, ask: in what ways can this step fail to produce its intended output. The output of step three is a list of failure modes phrased as observable process states, not effects on the product. For example, in a heat seal step, valid failure modes include "seal jaw temperature below setpoint," "seal jaw dwell time below setpoint," "film tension outside operating range," "missed seal." The downstream product effect (compromised seal integrity, contamination ingress, package failure during transport) goes in the next column.
Step 4: Map effects to severity
For each failure mode, list the effect on the end user, the patient if applicable, the downstream process, the customer, and the manufacturing operation. Rate Severity against the published AIAG-VDA table for the applicable FMEA type and the documented effect. Avoid generic number shortcuts that apply the same ratings across industries; the handbook descriptors reference impact categories (end user, regulatory, customer, plant) and the right number falls out of the descriptor that matches the actual effect.
Step 5: Identify causes and prevention controls
For each failure mode, identify the root causes. A useful structure is the 6M framework (man, machine, material, method, measurement, environment), but the categories matter less than the discipline of asking "what would have to be true for this failure mode to happen." For each cause, list the prevention controls that are already in place to keep it from occurring. Prevention controls are the engineering controls upstream of the failure: equipment design, mistake-proofing, supplier qualification, operator training, recipe lockdown.
Step 6: Rate occurrence
For each cause and its prevention control combination, rate Occurrence against the AIAG-VDA Occurrence table. The 2019 handbook revised the occurrence scale to reference defects per opportunity in similar processes rather than abstract probability. Use historical data when you have it. When you do not, document the assumption you made and what would have to be true for the rating to be wrong.
Step 7: Identify detection controls and rate detection
For each cause, list the detection controls that would catch the failure if prevention fails. Detection controls are typically in-process inspections, end-of-line tests, statistical process control limits, and downstream monitoring. Rate Detection against the AIAG-VDA Detection table, which ranks detection controls from "almost certain to detect" (1) to "no detection control" (10). Avoid fixed shortcuts such as "automated equals 2 to 3." The right rating depends on the actual control method, where in the process detection occurs, and whether the control detects the cause, the failure mode, or the effect. Document what the control detects and whether detection occurs before product release.
Step 8: Determine Action Priority
Apply the AIAG-VDA Action Priority table to the combined S, O, and D ratings to determine whether the failure mode is High, Medium, or Low priority. The Action Priority is not a calculation; it is a lookup. The handbook publishes the lookup table.
Step 9: Define and track recommended actions
For each High and Medium priority failure mode, define a recommended action that would reduce one or more of S, O, or D. Severity is reduced through design changes that eliminate or substantially mitigate the effect. Occurrence is reduced through stronger prevention controls. Detection is reduced through stronger detection controls. Assign an owner and a due date. Track actions in whatever system the organization uses for cross-functional corrective action; do not let actions live only inside the PFMEA spreadsheet.
Step 10: Re-rate after actions complete
When the action is complete and verified effective, update the S, O, and D ratings to reflect the new state, recompute Action Priority, and document the residual risk. The residual risk is the basis for the team's risk acceptance decision and the foundation of the PFMEA's defensibility in audit.
How PFMEA actually connects to validation
The connection between PFMEA and validation is one of the most underused leverage points in a regulated manufacturing quality system. Teams that get this connection right validate the right things at the right depth. Teams that get it wrong either over-validate (every parameter qualified, every step studied) or under-validate (qualifications that pass routinely but do not actually test the risks the process presents).
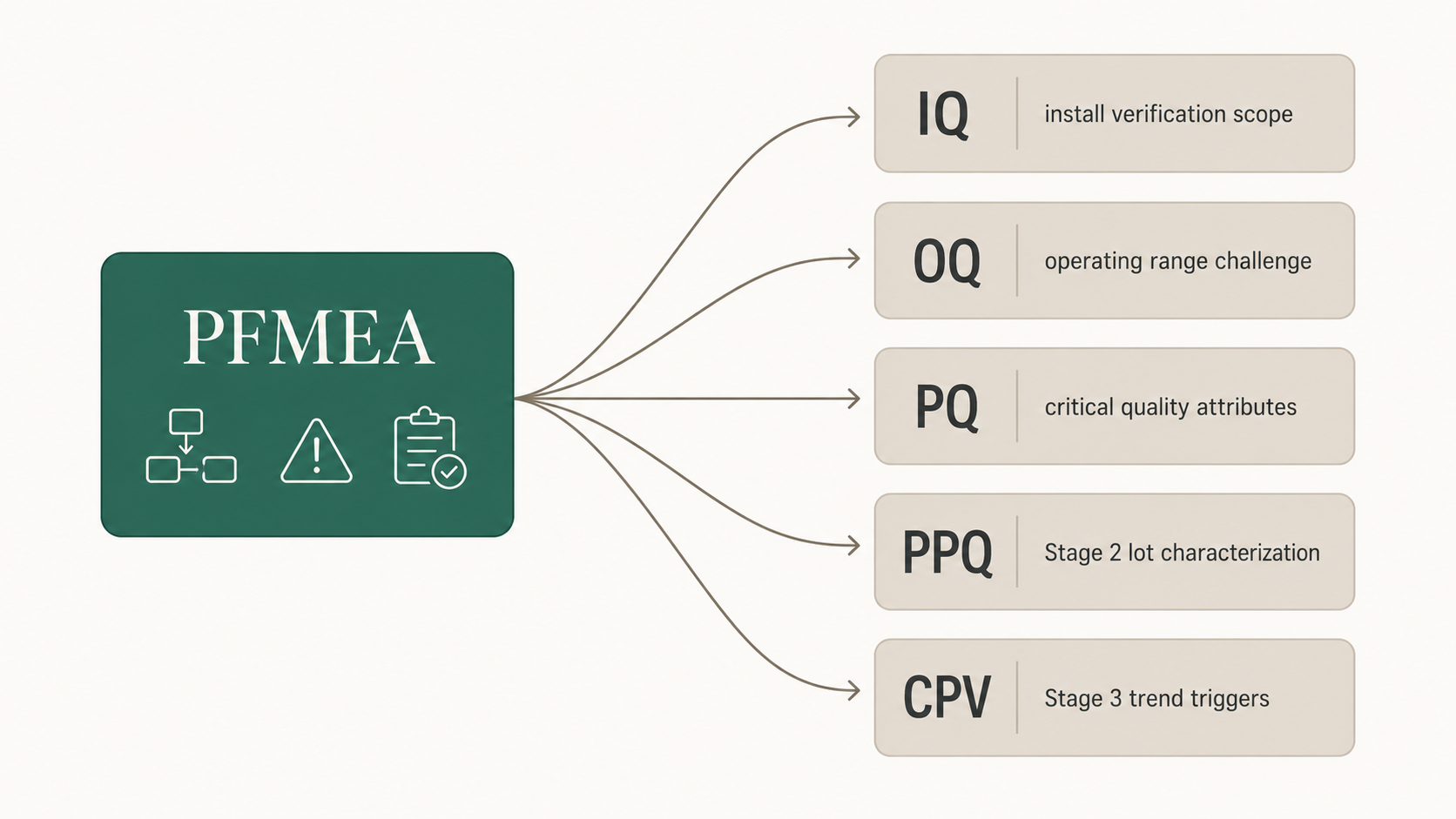
The mechanical link is this. The PFMEA identifies critical process parameters and the in-process controls that maintain them. The validation strategy then has to demonstrate that the equipment and process can hold those parameters within acceptable ranges under representative operating conditions. The PFMEA tells the validation team what to test; the validation protocols test it.
For an IQ protocol, the PFMEA informs which utilities, instruments, and equipment design features need installation verification. For an OQ protocol, the PFMEA informs which operating ranges and worst-case conditions need challenge. For a PQ protocol, the PFMEA informs which critical quality attributes have to be demonstrated under actual production conditions. For a PPQ at process validation Stage 2, the PFMEA informs which sources of variation have to be characterized across consecutive lots, and the PPQ protocol guide shows how those sources of variation become the sampling plan. For continued process verification at Stage 3, the PFMEA defines what shifts in process behavior would represent a meaningful change in the risk profile and trigger investigation.
Teams that skip this link end up writing OQ acceptance criteria anchored to vendor specifications rather than to process risk, and end up running PQ runs that demonstrate the equipment can do what the manufacturer says it can do, rather than that the process can hold the parameters PFMEA identified as critical. Auditors notice the difference. A risk-anchored validation strategy reads as a coherent program. A specification-anchored validation strategy reads as a checklist exercise.
The same link runs in reverse during change control. When a process change is proposed, the PFMEA is the document that lets the team assess whether the change shifts S, O, or D for any failure mode and therefore whether revalidation is needed. A PFMEA that is current is the fastest path through a change review. A PFMEA that is stale forces every change review to re-derive the risk picture from scratch.
PFMEA mistakes that flag during audits
Auditors do not read every row of a PFMEA. They sample. The patterns that draw their attention are recognizable.
The first pattern is high-severity failure modes with no documented action and no residual risk acceptance. A Severity 9 row sitting at Action Priority High with the recommended action field blank tells the auditor that either the rating is wrong, the action was never opened, or the team is hoping no one notices. Any of those reads badly.
The second pattern is RPN-only documentation. A PFMEA dated after 2019 that still uses RPN exclusively, with no Action Priority column and no AIAG-VDA reference, signals that the team has not refreshed against current standards. The same pattern shows up as a PFMEA template that pre-dates the team's QMSR transition for medical device manufacturers.
The third pattern is unchanged ratings over multiple revisions. A PFMEA that has been revised three times in five years with no rating changes signals a paper exercise. Real processes change. Real PFMEAs reflect those changes.
The fourth pattern is detection controls that double-count the same inspection. A failure mode listed with Detection 2 because "QC inspects 100 percent of finished product" reads as defensible until the auditor finds the same QC inspection cited as the Detection control for fifteen other unrelated failure modes. Detection controls have to be specific to the failure mode in question.
The fifth pattern is missing linkage to deviations. When the deviation system records a process failure that matches a PFMEA failure mode, the PFMEA should have been updated to reflect that the failure mode actually occurred. A deviation log showing a recurring failure mode and a PFMEA that still rates that mode at Occurrence 2 is a documentation gap an auditor will pursue.
The sixth pattern is PFMEAs that have never been re-rated after corrective action. A high-priority failure mode that drove a major CAPA two years ago and still shows the original S, O, D ratings tells the auditor that the team treated the CAPA as a checkbox rather than a residual risk update.
The seventh pattern is missing or thin cross-functional review. A PFMEA signed only by manufacturing engineering, with no quality or operations signature, signals that the analysis is one team's view of the process rather than the organization's view. AIAG-VDA explicitly calls for cross-functional team participation.
Living document: when to update
A PFMEA is updated when the process it describes changes. The triggers are similar to the revalidation triggers for IQ, OQ, and PQ, with a few PFMEA-specific additions.
Equipment changes that alter how a process step is executed trigger a PFMEA update. Recipe or parameter changes that alter setpoint or tolerance trigger a PFMEA update. Material changes that introduce a new failure mode or change the occurrence of an existing one trigger a PFMEA update. Supplier changes that affect incoming material variability trigger an update. Software or firmware updates on equipment that runs the process trigger an update if the update changes how the process is controlled or monitored. New failure modes observed in production, in deviation investigations, or in field complaints trigger an update. Regulatory standard updates that change required risk management practice trigger an update.
A useful operational rule is that the PFMEA gets reviewed for currency at the same cadence as the Validation Master Plan: annually at minimum, plus on every change control that touches the process under analysis. Annual review without a recent process change typically results in no rating changes but does produce a documented confirmation that the team has looked.
Closing
PFMEA earns its place in a regulated manufacturing quality system when it drives decisions rather than documents them after the fact. The teams whose PFMEAs survive audit scrutiny are the teams whose validation scope, in-process controls, change reviews, and CAPAs reference the PFMEA as the source of truth for process risk. The teams whose PFMEAs flag during audits are the teams whose PFMEAs sit in a folder, updated only when a regulator forces the issue.
The mechanics under AIAG-VDA are not complicated. Define the process boundary. Decompose into steps. Identify failure modes. Map to effects. Rate Severity, Occurrence, and Detection against the published rubrics. Look up Action Priority. Define actions for High and Medium priority modes. Re-rate after action. Update on change. The discipline is in keeping the document connected to the work the organization actually does.
---
Valiqa is an AI-powered validation lifecycle platform for regulated manufacturing. Learn more at valiqa.io
Frequently Asked Questions
Ready to automate your validation documentation?
Generate audit-ready IQ/OQ/PQ protocols in minutes, not weeks.
Get StartedWe use essential cookies for authentication and security. With your consent, we also use Microsoft Clarity, Google Analytics, and the LinkedIn Insight Tag on our marketing pages to understand how visitors navigate the site and to measure our advertising. Learn more.